Getting a generic medicine approved in the European Union used to feel like trying to solve a puzzle where the pieces kept changing shape. You had one set of rules for Germany, another for France, and yet another for Poland. But if you are looking at the landscape today, specifically after the major shifts brought by the EU Pharma Package finalized in mid-2025, the game has changed significantly.
The system is still complex, but it is more predictable than it was five years ago. The core goal remains the same: balance patient safety with market competition. Generics currently make up about 65% of all prescriptions in the EU by volume, according to data from the European Generic and Biosimilar Medicines Association (EGA). However, they only account for 18% of the total value. This gap highlights why manufacturers fight so hard for efficient approvals-speed to market is everything.
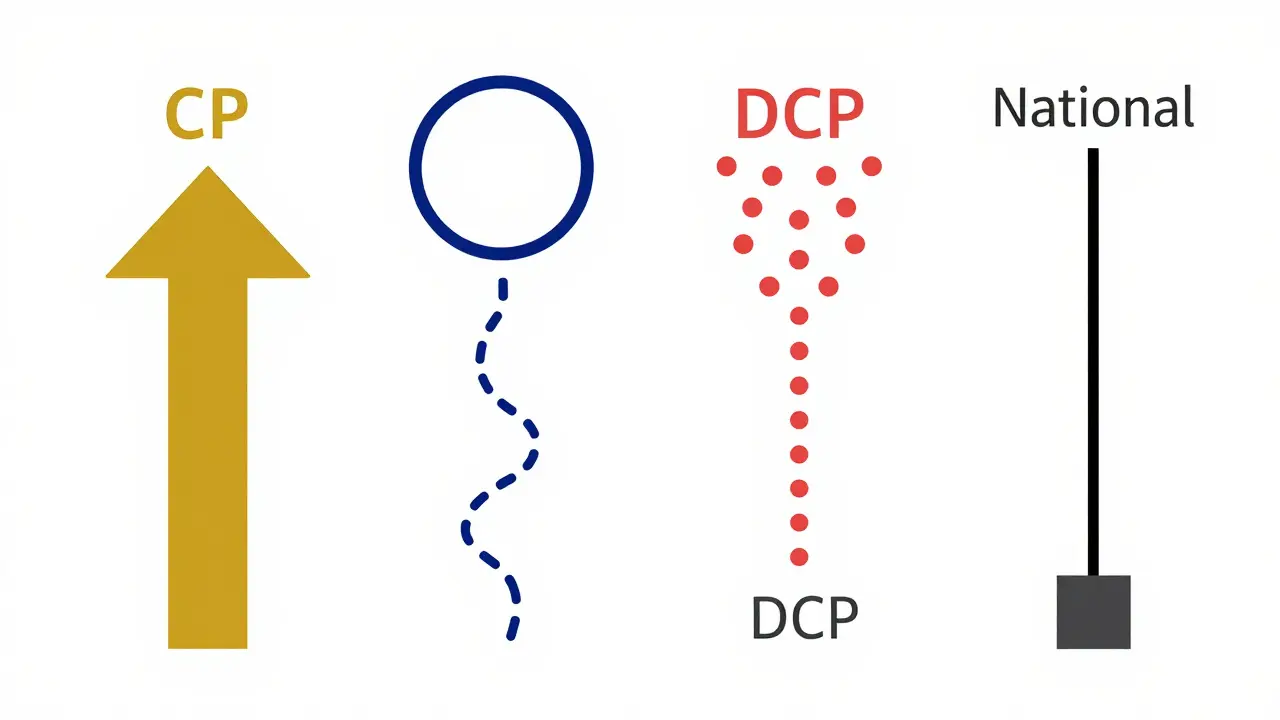
Understanding the Four Approval Pathways
To enter the European market, you cannot just pick any route. The regulatory framework offers four distinct pathways, each with its own speed, cost, and strategic implications. Choosing the wrong one can add months-or even years-to your launch timeline.
- Centralized Procedure (CP): This is the golden ticket for many. You submit one application to the European Medicines Agency (EMA), and if approved, you get marketing authorization valid across all 27 EU member states plus Iceland, Liechtenstein, and Norway. It’s used for about 15% of new generic applications. Under the 2025 reforms, the scientific assessment time has been tightened to 180 days, down from 210. The European Commission then grants authorization within 46 days. It’s fast, but expensive-expect application fees around €425,000 plus consultancy costs ranging from €1.2 to €1.8 million.
- Mutual Recognition Procedure (MRP): Used for roughly 42% of generics, this allows you to get approved in one country (the Reference Member State) and then seek recognition in others. Ideally, consensus should be reached in 90 days. In reality, a 2024 IQVIA analysis showed the average time is closer to 132.7 days due to divergent national requirements. Costs are lower, typically €180,000-€220,000, but you face sequential risks. If Germany delays pricing negotiations, your entry into the Netherlands or Belgium gets stuck too.
- Decentralized Procedure (DCP): This accounts for 38% of applications. You submit simultaneously to multiple countries without prior national approval. A Reference Member State leads the assessment. While the clock says 210 days, coordination challenges often push the average to 247 days. It’s notoriously problematic, with 37% of applications facing delays over six months, particularly in Eastern European markets where quality requirement interpretations vary wildly.
- National Procedure: Only 5% of applications use this. You go straight to a single member state’s authority. It takes 180-240 days and limits you to that one market. It’s rarely strategic unless you are targeting a specific high-reimbursement niche, but even then, it fails to leverage EU harmonization.
The Impact of the 2025 Pharma Package Reforms
The reforms that took effect in 2025 are not just tweaks; they are structural changes designed to fix the fragmentation that plagued the previous decade. The most immediate benefit for generic manufacturers is the expansion of the Bolar exemption.
Previously, you could only start pricing and reimbursement negotiations two months before patent expiry. Now, you have a six-month window. According to a 2025 economic model by REMAP Consulting, this simple change accelerates market entry by an average of 4.3 months. That’s nearly half a year of head start on competitors who aren’t prepared.
Another major shift involves Regulatory Data Protection. The standard period has been reduced from 10 years to 9 years (an 8+1 structure). This can extend to 10 years if public health targets are met, but the baseline reduction creates a more dynamic market. Dr. Sabine Rödl, Director of EGA, noted that this preserves innovation incentives while allowing generics to enter sooner. However, there is a catch: the introduction of Transferable Exclusivity Vouchers tied to a €490 million sales threshold might disadvantage mid-sized firms that don’t hit those revenue marks.
Bioequivalence: The Non-Negotiable Standard
Regardless of which pathway you choose, the science must hold up. You must prove that your generic is therapeutically equivalent to the reference product. This means identical qualitative and quantitative composition of active substances, the same pharmaceutical form, and proven bioequivalence.
The EMA’s Guideline on the Investigation of Bioequivalence requires rigorous studies. Your 90% confidence intervals for Cmax (maximum concentration) and AUC (area under the curve) must fall strictly within 80.00-125.00%. There is no wiggle room here.
However, "identical" doesn’t always mean straightforward. Complex generics, such as inhalers or modified-release formulations, face extra scrutiny. For instance, Germany’s BfArM often demands additional pharmacodynamic studies beyond standard EMA requirements. A 2025 survey by the Association of the British Pharmaceutical Industry (ABPI) found that 68% of generic companies cited inconsistent national bioequivalence requirements as their top hurdle. If you are launching a complex generic, expect to spend 15-18 months preparing documentation alone, including 6-8 months dedicated to comparative studies.
| Pathway | Market Coverage | Avg. Timeline | Est. Cost Range | Best For |
|---|---|---|---|---|
| Centralized (CP) | All 27 EU + EEA | ~226 days | €1.6M - €2.2M+ | High-value, pan-EU launches |
| Mutual Recognition (MRP) | Multiple (Sequential) | ~133 days | €180k - €220k | Mid-tier products, flexible entry |
| Decentralized (DCP) | Multiple (Simultaneous) | ~247 days | Variable | Regional strategies, lower risk |
| National | Single Country | 180-240 days | Lowest | Niche, high-reimbursement markets |

Operational Realities and Hidden Costs
It’s not just about getting the stamp of approval. It’s about what happens next. The 2025 reforms introduced mandatory electronic product information (ePI) submissions in XML format by 2026. This isn’t a minor IT update. White & Case estimates that upgrading infrastructure for this compliance will cost companies between €180,000 and €250,000.
Supply chain planning is also becoming tighter. The new "obligation to supply" mechanism aims to reduce medicine shortages, projected to drop them by 35% by 2028. But Professor Panos Kanavos of LSE Health warns that national authorities interpret "sufficient quantities" differently. In smaller markets, this could create artificial supply constraints that delay your actual availability on pharmacy shelves, even if you have legal approval.
Consider the experience of Sandoz with its generic version of Cosentyx. By leveraging the Centralized Procedure, they achieved a simultaneous EU-wide launch in Q2 2025-11 months faster than a traditional MRP approach would have allowed. Conversely, Teva faced an 8.2-month delay in Dutch and Belgian markets for generic rosuvastatin because German pricing negotiations stalled the MRP process. These real-world examples show that pathway selection is a financial decision, not just a regulatory one.
Market Context and Future Outlook
The European generics sector was valued at €42.7 billion in 2024, showing a 6.2% growth rate. Central and Eastern Europe are growing fastest, at 9.8% annually, according to IQVIA’s 2025 report. This regional disparity means your strategy might need to differ depending on whether you are targeting Western Europe or emerging markets in the East.
Competition is intensifying. Indian manufacturers captured 38% of EU generic approvals in 2024, up from 29% in 2020. European firms like Sandoz and Viatris maintain a 52% market share, largely by dominating the Centralized Procedure. As the revised Regulatory Data Protection framework fully implements on July 1, 2026, we expect accelerated entry for 78 high-value biologics currently in development. Evaluate Pharma projects that these reforms will increase the generic prescription share to 69.2% by 2028.
If you are navigating this space, remember that harmony is still a work in progress. National peculiarities remain-France requires specific pediatric formulation documentation, and Germany demands extra stability data for polymorphic compounds. Stay agile, invest in digital compliance early, and choose your pathway based on your target market’s reimbursement landscape, not just the speed of approval.
What is the biggest change in the 2025 EU Pharma Package for generics?
The most significant operational change is the expansion of the Bolar exemption, allowing manufacturers to begin pricing and reimbursement negotiations six months before patent expiry instead of just two. Additionally, the standard Regulatory Data Protection period has been reduced from 10 years to 9 years (8+1), potentially accelerating market entry for many products.
Which approval pathway is fastest for EU-wide coverage?
The Centralized Procedure (CP) via the EMA is the fastest for pan-European coverage. With the 2025 reforms, scientific assessment takes 180 days and European Commission authorization takes 46 days. While it has higher upfront costs, it avoids the sequential delays associated with Mutual Recognition or Decentralized Procedures.
How much does it cost to submit a Centralized Procedure application?
Application fees are approximately €425,000. However, when you include necessary consultancy and preparation costs, the total investment typically ranges between €1.2 million and €1.8 million. This makes CP viable primarily for high-value generics with projected EU-wide sales exceeding €250 million annually.
What are the bioequivalence requirements for EU generics?
Generics must demonstrate identical qualitative and quantitative composition of active substances and the same pharmaceutical form as the reference product. Bioequivalence studies must show that 90% confidence intervals for Cmax and AUC fall within the strict range of 80.00-125.00%.
When do the new ePI submission requirements take effect?
Mandatory electronic product information (ePI) submissions in XML format are required by 2026. Companies are advised to start investing in IT infrastructure now, as compliance upgrades are estimated to cost between €180,000 and €250,000 per company.